Alkaptonuria
Skin Conditions
Alkaptonuria is a genetic disorder that is inherited in an autosomal recessive manner, which means that both parents of the patient must (with a few rare exceptions) be either asymptomatic carriers of this disease or have the disease themselves. Alkaptonuria is a result of a mutation that causes a protein/enzyme (homogentisate 1,2-dioxygenase) to become defective. This protein, when functioning properly, is integral in the breakdown of the amino acids tyrosine and phenylalanine. Without this enzyme one of the intermediates in this breakdown pathway, homogentisic acid, accumulates to abnormal levels in the body and interferes with normal bodily functions resulting in the symptoms associated with alkaptonuria.

Alkaptonuria
Skin Conditions
Alkaptonuria is a genetic disorder that is inherited in an autosomal recessive manner, which means that both parents of the patient must (with a few rare exceptions) be either asymptomatic carriers of this disease or have the disease themselves. Alkaptonuria is a result of a mutation that causes a protein/enzyme (homogentisate 1,2-dioxygenase) to become defective. This protein, when functioning properly, is integral in the breakdown of the amino acids tyrosine and phenylalanine. Without this enzyme one of the intermediates in this breakdown pathway, homogentisic acid, accumulates to abnormal levels in the body and interferes with normal bodily functions resulting in the symptoms associated with alkaptonuria.
Alkaptonuria Symptoms
The classic symptom of alkaptonuria is urine that darkens shortly after it leaves the body. This is a result of oxidation of the homogentisic acid that is found in the urine of these patients. The excess homogentisic acid that is not excreted in the urine is first oxidized to benzoquinone acetic acid and then slowly deposited into the patient’s collagen (a major component of cartilage and connective tissue) resulting in a progressive darkening to blue or grey of these structures over time.
These changes in the cartilage (also known as ochronosis) do not typically become apparent until middle age, at which point external pigment changes appear, especially in the ear (where cartilage is just below the skin) and the whites of the eyes (vision is unaffected). Another more debilitating manifestation of alkaptonuria is early onset arthritis due to deposition of benzoquinone acetic acid in joint cartilage, especially in the spine – in many patients the first symptom of their disease is lower back pain. In some cases deposition in the cartilage of the heart valves can result in inflammation of these structures leading to heart disease.
Benzoquinone acetic acid may also collect in the cartilage of the trachea and lungs resulting in breathing problems. Finally, individuals with alkaptonuria are at increased risk for developing stones in the kidneys, gallbladder, salivary glands, and prostate, and for bone fractures.
Alkaptonuria Diagnosis and Treatment
Unfortunately, there is no cure for alkaptonuria – most treatment relies on prevention and treatment of the symptoms listed above. Despite the many potential manifestations of this disease there is no negative impact on life expectancy, though quality of life may be reduced. A low protein diet is thought to help delay development of this disease due to reduced intake of the amino acids tyrosine and phenylalanine – the precursors of homogentisic acid. Some physicians may utilize large doses of vitamin C to slow the conversion of homogentisic acid to benzoquinone acetic acid, though the benefit of this treatment had been disputed.
Recent research has revealed that nitisinone, a drug that has been used to treat a different disease involving tyrosine metabolism, can inhibit the conversion of tyrosine to homogentisic acid thus greatly reducing the levels of this particle in the body. The long-term effects of this drug are unknown. Patients with alkaponturia often receive joint replacement surgery to treat their arthritis and heart surgery to correct valve defects. Other manifestations are treated as they arise.
Alkaptonuria is most often diagnosed in middle age as the painful symptoms of this disease begin to impinge on daily activities. However, the earlier it is detected the better the prognosis. If you or your child begins to experience signs or symptoms of alkaptonuria – especially urine that darkens significantly after exposure to air as this is often one of the earlier symptoms – schedule an appointment with your physician for evaluation and treatment.
Related Medical Dermatology Articles

Welcome Phi-Yen “Mimi” VuTran, PA-C. Mimi is the first physician assistant to work with Contour Dermatology. Voted Best of the Desert, Best PA in 2022.
Dr. Timothy Jochen extracts a lipoma that looks like a lobster! Watch as this huge lipoma is skillfully extracted and the would is sutured by the surgeon.
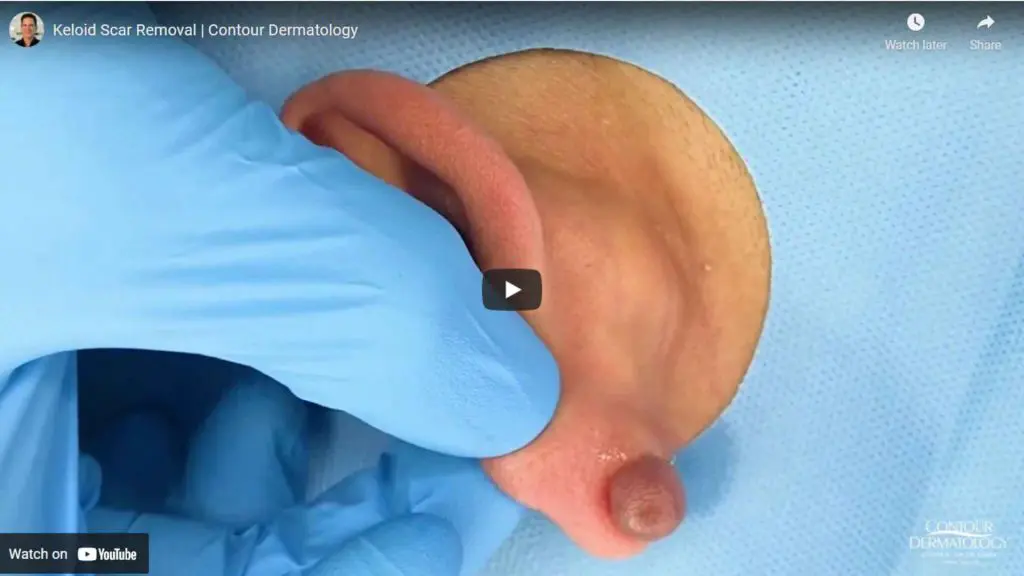
Dr. Jochen removes a huge ear keloid caused by an earring. Keloids are scar tissue that has expanded beyond the boundaries of the original wound.
The hair loss of alopecia areata, skin depigmentation of vitiligo and skin lesions of psoriasis are conditions that affect a person’s body but their wellbeing.

























